Basic research
For the past 14 years, the primary goal of Dr. Beth Roman’s research has been to better understand why AVMs develop when ALK1 signaling is impaired. The model system that her laboratory uses is the zebrafish, a small freshwater fish that some of you might have in your home aquaria. The zebrafish embryonic vascular system develops very similarly to the human vascular system, with the same biochemical signals directing the assembly of endothelial cells (the cells that line the inner surface of blood vessels) into a correctly patterned network of interconnected tubes. Because zebrafish embryos are fertilized outside of the mother, are optically transparent, and are fast-developing, Dr. Roman’s team can noninvasively watch these tubes form in real time in live animals.
The Roman lab studies a strain of zebrafish that harbors a mutation in ALK1, the same gene that is mutated in patients with HHT2. Zebrafish embryos that have two copies of this ALK1 mutation develop improper connections between the major arterial and venous blood vessels in the head: in short, they form AVMs at a predictable time in a predictable location. Using sophisticated microscopes, the Roman lab can capture the endothelial cell movements that give rise to AVMs in live zebrafish embryos, thereby leading to a better understanding of the natural history of these vascular lesions.
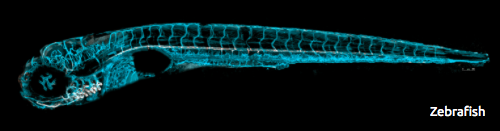
Using sophisticated microscopes, the Roman lab can capture the endothelial cell movements that give rise to AVMs in live zebrafish embryos, thereby leading to a better understanding of the natural history of these vascular lesions.
Using this zebrafish ALK1 mutant model, the Roman lab has discovered that AVM development is not the direct result of defective ALK1 function. In this model, the arteries that normally express ALK1 enlarge, primarily because they contain too many endothelial cells. Because these arteries enlarge, the amount of blood flowing through them increases, and the mechanical forces that the blood vessels are exposed to increase in magnitude. This can be a dangerous situation, so blood vessels have evolved strategies to keep mechanical forces in check. By studying zebrafish ALK1 mutants, the Roman lab has discovered that one strategy used by blood vessels to try to normalize mechanical force is to allow blood to drain from enlarged arteries through what would normally be temporary, primitive connections to neighboring veins. In wild type embryos, these connections exist for less than one day, disappearing as the developing vasculature remodels and matures. In ALK1 mutant embryos, these connections are retained and enlarged, giving rise to a permanent arterial-venous shunt, or AVM. In short, these findings suggest that a “normal” adaptive response to the change in hemodynamic force downstream of enlarged arteries is what ultimately causes AVMs in this model of HHT2. The Roman lab is currently following up on these findings by asking why more endothelial cells wind up in ALK1-dependent arteries when ALK1 is not functioning, and by studying how mechanical force is transformed into a biochemical signal that leads to force-dependent development of AVMs.
Translational research
Another major focus of the Roman lab is to discover drugs that may help prevent AVMs and telangiectasias in HHT patients. HHT is a haploinsufficiency: endothelial cells that produce only half of the “normal” amount of ALK1, ENG, or SMAD4 cannot consistently maintain proper connections between arteries and veins. Therefore, drugs that increase expression of these genes or activity of the ALK1/ENG/SMAD4 pathway could be useful as HHT therapeutics. The Roman lab is taking a two-pronged approach to this problem. First, they are attempting to better understand how ALK1 “signals” in endothelial cells by identifying proteins that act both upstream and downstream of ALK1 itself. For example, the team recently identified bone morphogenetic protein 10 (BMP10) as a key upstream activator of ALK1 in embryonic blood vessels. Identifying additional players in this pathway will open access points for development of targeted therapeutics that enhance flux through this signaling pathway. Second, using a zebrafish line engineered in the Roman lab to express an easily visible green fluorescent protein that serves as a faithful “readout” of arterial ALK1 expression, the research team is testing a large number of chemicals for their ability to increase ALK1 expression. This second, unbiased approach does not rely on pathway knowledge and is therefore complementary to the first approach.
Research Publications
Disruption of acvrl1 increases endothelial cell number in zebrafish cranial vessels
Roman, B.L., Pham, V.N., Lawson, N.D., Kulik, M., Childs, S., Lekven, A.C., Garrity, D.M., Moon, R.T., Fishman, M.C., Lechleider, R.J., and Weinstein, B.M.
ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2
Park, S.O, Lee, Y.J., Seki, T., Hong, K.H., Fleiss, N., Jiang, Z., Park, A., Wu, X., Kaartinen, V., Roman, B.L., and Oh, S.P.
Interaction between alk1 and blood flow in the development of arteriovenous malformations
Corti, P., Young, S., Chen, C.Y., Patrick, M.J., Rochon, E.R., Pekkan, K., and Roman, B.L.
Circulating Bmp10 acts through endothelial Alk1 to mediate flow-dependent arterial quiescence
Laux, D.W., Young, S., Donovan J.P., Mansfield, C.J., Upton, P.D., and Roman, B.L.
BMP9 Mutations Cause a Vascular-Anomaly Syndrome with Phenotypic Overlap with Hereditary Hemorrhagic Telangiectasia
Wooderchuk-Donahue, W.L., McDonald, J., O’Fallon, B., Upton, P.D., Li, W., Roman, B.L., Young, S., Plant, P., Fulop, G., Langa, C., Morrell, N.W., Botella, L.M., Bernabeu, C., Stevenson, D.A., Runo, J.R., and Bayrak-Toydemir P.
Genetic and molecular basis for hereditary hemorrhagic telangiectasia
Roman, B.L. And Finegold, D.N.
Context-specific interactions between Notch and ALK1 cannot explain ALK1-associated arteriovenous malformations
Rochon, E.R., Wright, D.S., Schubert, M.M., and Roman, B.L